Sepsis
This is a review of the current Sepsis guidelines and then a look at the pathophysiology of sepsis. We will then look at the ideas behind treating sepsis and the complications of it whilst taking a critical look at the guidelines and try and work out where they could be lacking – hopefully bridging the gap between the protocolized treatment of infection and sepsis and the deeper understanding we are hopefully going to build. To avoid over complicating we will focus on adult care.
Throughout this page there are hyperlinks that will take you to the relevant paper or guideline that is being discussed or referenced.
Sepsis Summary - if you want just the brief overview.
The pathophysiology of sepsis can be summarised as an excessive inflammatory response to endotoxins, mediated by inflammatory cytokines. The process causes damage to the endothelial glycocalyx, leading to a distributive, vasodilatory shock. The end result of this is hypotension and tachycardia with organ failures following on from there.
Our initial treatment should be source control and management – reduction and removal of the cause of the inflammation. This boils down to antibiotics and surgical removal of the inflammatory source where suitable (I+D of collections/abscess, resection of dead/dying bowel etc etc)
Fluid forms the main part of the initial resuscitative measures. The aim is restoring the circulating volume but too much can be harmful to the glycocalyx and lead to complications such as fluid overload and ARDS. The practice is 30ml/kg and then consideration of vasopressors (not inotropes)
The most popular vasopressor is Noradrenaline, which is usually given centrally with arterial line monitoring. It can be used in conjunction with vasopressin and adrenaline as the situation deteriorates. Other ‘pressors such as phenylephrine and metaraminol can be used as temporising measures whilst establishing norad.
Adjuncts to sepsis management are typically not evidenced based but are often used as they make logical sense and have been shown to cause no harm. They are Corticosteroids and Bicarbonate. They tend to be used in severe or refractory cases. Others such as insulin and methylene blue are more controversial and tend not be used.
Finally treatments of the complications of sepsis, like renal failure and ARDS, haven’t been covered and are topics more of a critical care perspective.
The important background for this is the climate in which the guidelines have been developed – there has been identification of a lot of preventable deaths where “sepsis” has been missed or the treatment has been delayed.
There are approximately 200000 cases of sepsis in the UK each year with around 48000 deaths. But this is based on coding data and is thought to be a low estimate – there are 1.5million patients suffering from severe infection in England each year and there maybe as many as 70000 deaths a year.
Have a look at the patient case below and we work through what we need to know to optimally understand and treat what is going on.
You are asked to review a 47-year-old male in the Emergency Department with hypotension that has not responded to rapid infusion of 2 litres intravenous crystalloid. On examination ahis temperature is 40°C, he is warm peripherally with a respiratory rate of 24 breaths per minute, an arterial oxygen saturation of 98% on room air, a heart rate of 140 beats per minute, and a blood pressure of 80/40 mmHg with an arterial metabolic acidosis and a lactate concentration of 6 mmol/L.
1. Current NICE Sepsis Guidelines
NICE has moved to a sepsis risk stratification tool dependent on age. It suggest that we should be thinking “could this be sepsis?” in any patient with a suspected infection.
It then breaks it down to low, moderate and high risk criteria based on physiological parameters and clinical evidence of deranged physiology.
The areas that it looks at are HR, RR, BP as the physiological parameters and then cognition, urine output and clinical evidence of shock (mottling) or poor perfusion (cyanosis).
It then recommends treatment based on the criteria – the more deranged the physiology the more prompt and thorough the response it recommends. Essentially boiling the treatment down to fluid resuscitation and early antibiotics with senior involvement ASAP.
If we were going to make a definition from this I’d suggest it would be an infection causing derangement in physiological parameters and function. But the move away from a definition reflects the reality of the confounding features behind making the diagnosis and what we are really interested in is recognizing and treating unwell patients successfully.
NICE also suggests that we should be thinking about patients that are higher risk of getting sepsis and treating them with more caution. The list is of people with impaired ability to combat infection and so mount a strong physiological response and/or those who are more frail and vulnerable to infections:
- Young (<1yr) and Old (>75yrs)
- Immunosuppressed
o Neutropenic – both literal (post chemotherapy) or functionally (blood cancers)
o Suppressed function (diabetics, splenectomy, sickle cell)
o Long term steroids
o DMARDs
- People who have recently had a physiological insult like those who have had a recent operation
- People with compromised protections from infection, like those with breaches in their skin, IVDUs or have indwelling lines or catheters
2. Physiology behind Sepsis
So how does the deranged physiology occur from an infection – giving us sepsis.
Generally speaking it is a systemic inflammatory response from a stimulus like a bacterial toxin. The pro-inflammatory mediators thought to be involved are Tumour necrosis factor and interleukin-1 (cytokines). The response to these cytokines can explain most of the problems we see clinically with sepsis. Interestingly they seem to cause neutrophil-endothelial cell adhesion, active the clotting mechanism and generate microthrombi, seemingly related to degradation of the glycocalyx which would otherwise inhibit this effect.
The initial inflammatory response is to enable delivery of necessary to components to the infected are - WBC, fibrin and platelets. The process doesn’t discriminate between injury and infection, it is just an inflammatory response
First up is a hypermetabolic phase of the infection/stimulus leading to a vasodilation and an increase in cardiac output – it may only be demonstrated clinically though more subtle markers of physiological derangement – an increased HR that doesn’t necessarily trigger scoring systems or a RR rise reflecting the increased O2 demand.
As our compensatory mechanisms fail and start to become counterproductive we move into a physiological state we are more familiar with – that of septic shock and organ dysfunction.
A summary is in the diagram below. But I think you’ll agree we need to go into it a little deeper and can probably add extra knowledge to put some meat on these bones. We can look at each of the organ failures associated with sepsis in turn


Hypoxic Respiratory Failure
So, without commenting on specific infections (eg pneumonia) that may make you hypoxic, sepsis as a whole affect your ability to deliver O2 to your tissues – and as we know this is the “big picture” of a lot of our physiology and what we try to maintain in a lot of emergency and critical care.
Essentially the physiology of hypoxia is the increased whole body O2 demand combined with lung injury from generalized inflammation causing vasodilation and therefore leaky capillaries, increasing the diffusion distance and causing ARDS. The discrepancy of the increase demand and impaired functioning leads to the hypoxic respiratory failure.
Haemodynamic Collapse and Shock
Two main drivers behind this – impaired cardiac function and loss of systemic vascular resistance
1. Impaired cardiac function can be caused by multiple things, firstly direct toxin effect may reduced myocardial contractility but more commonly metabolic disturbances like a profound metabolic acidosis or electrolyte disturbances will impede the heart to a greater effect
2. The main defining feature of Septic Shock is the circulatory collapse from the loss of peripheral vascular resistance.
This is driven by the systemic inflammatory response causing vasodilation and damage to the endothelial glycocalyx leading to “leaky capillaries”

It’s a little bit of an aside but an important one. What is the endothelial glycocalyx?
- A hydrated gel layer of glycoproteins, polysaccharides and proteoglycans
- Lies between the vessel wall and the blood, forming an interface.
- Has the main role when it come to transvascular fluid exchange
The glycocalyx is damaged by hyperglycaemia, hyperlipidaemia, smoking, inflammation and aggressive fluid resuscitation
- The theory behind it is that it is maintained by the contents of plasma so can quickly repair itself under normal circumstances and so despite it being very fragile it can repair quickly – there is lab evidence of it repairing in under a second when provided with a temporary injury
- In forming the interface between the moving blood and the vessel wall it take on number or important functions
o Limits blood cell interaction with the endothelium
o Stops fluid, protein and lipid leakage across the vascular wall
§ Maintaining oncotic pressure
o Limits the interaction of inflammatory cells and platelets to the endothelial surface
o Mediates the shear stress dependent nitric oxide production by acting as a mechanotransducer of fluid shear force
o Retains protective enzymes and anticoagulation factors
- So what happens in the context of sepsis
o Inflammatory response and resuscitation hypervolaemia lead to glycocalyx break down (TNFalpha), leading to:
§ Capillary leak – tissue and organ oedema, impaired microcirculatory oxygen delivery and so impaired organ function
§ Increased platelet activation and local hypercoagulability – local mircothrombosis leads to microvascular shunting – impaired perfusion and increased lactate production
· This could lead to DIC
§ Loss of vascular responsiveness – vasoplegia and hypotension
- What can we do to look after the glycocalyx if its so important? There are 3 accepted measures.
o Corticosteroids
o Normalglycaemia
o Normovolaemia (ie don’t overload them)
Acute Renal Failure
- Pre-renal AKIs are a common consequence of sepsis and monitoring urine output is a significant part of treating unwell patients with shock or acidosis to demonstrate organ functioning. The other clinical marker of this that we can directly observe is the cognition of the patient – significant toxins or hypoperfusion of the brain will lead to reduced GCS and cognitive impairment, commonly seen in the elderly as a hypoactive delirium which is often an acute on chronic cognitive impairment related to minimal physiological insult.
- There is also evidence for tubular dysfunction secondary to inflammatory cytokines which will be unrelated to hypotension.
Hepatic Dysfunction
- Total hepatic failure is uncommon in sepsis, but it is common to see LFT derangement and synthetic impairment suggesting an element of hepatic injury, often from hypoperfusion
- Any impairment of hepatic function will, over time, contribute to hypalbuminaemia and coagulopathy
Septic Encephalopathy
- The mechanism behind this is poorly understood but seems to be related to microvascular damage and impaired mitochondrial oxygenation in combination with direct action of cytokines.
- What is established is that diffuse cerebral dysfunction in sepsis is often associated with poor prognosis
Coagulopathy
- Given the depletion and use of coag factors in the microvascular changes and glycocalyx there is often a subclinical coagulopathy in the absence of DIC
- There is also reduced hepatic production of coag factors that contributes to the coagulopathy
As you can see, the move away from defining sepsis by SIRS, Sepsis-3 or by a SOFA score has been a good one – it is much more multifaceted and varied than those scoring systems allow for. They were neither sensitive or specific leading to their use becoming more academic than clinical.
Now if we think about our clinical case – the 40yo – he is clearly infected with evidence of deranged physiology both in his parameters and biochemically with that raised lactate and metabolic acidosis.
If we look at the NICE criteria or at our own understanding of sepsis he clearly is “high risk” or sick sick. Now the question becomes what are the treatment options?
3. Management of Sepsis
Now the easy answer here is the sepsis 6. It has been around forever (it was even a thing when I was at med school and we believed in hypoxic drive) and remains a good starting point. Take 3, Give 3
Take - Blood Cultures, Lactate, Urine Output
Give - Antibiotics, Oxygen (if needed), Fluids
An alternative sepsis 6 to the one I learnt at med school is a little more in depth in describing the basics – this has been taken directly from the sepsis trust’s manual

But we aren’t here for the basics – we set out a lot of questions to answer in how to manage septic patients and if the 500ml bolus recommended in the NICE guidelines doesn’t get us out of trouble, or as the Sepsis 6 from the Sepsis Trust says – you are the senior clinician, what do we do next?
Lets start by breaking down the problems caused by sepsis – those of increase O2 demand and hypoxia, shock, Cardiac Dysfunction, Hepatic Failure, Renal Failure and coagulopathy.
The initial problem of increased O2 demand and hypoxia is easily resolved – give enough O2 to maintain sats in an appropriate range, without over oxygenating.
Most of the remaining problems can be explained by the haemodynamic compromise caused in sepsis (shock, hepatic and renal failure) so let’s start there.
The cause of haemodynamic compromise is a combination of vasodilation and fluid distribution out of the intravascular space.
- The initial management of this is simple – IV fluid to replace the redistribution in the intravascular space, but also to account for any reduced oral intake whilst unwell and the increased insensible losses from being hypermetabolic and pyrexial.
- However, we don’t want to give too much, as we have already mentioned too much fluid leads to further glycocalyx damage, exacerbating the distributive effect. Too much fluid will also lead to oedema in tissues – increasing diffusion distances , contributing to end tissue hypoxia and an ARDS picture in the lungs
- So what’s the accepted practice? What were our clinical questions?
o Are fluids the answer?
§ If so what?
· Colloids?
· Balanced Crystalloids?
· Saline?
§ How much?
- Our options are colloids vs saline vs balanced crystalloids
o Now colloids have been out of fashion for a long time – they were shown to be harmful – and this fits with our greater understanding of the endothelial glycocalyx that dovetailed with these results. In theory the increased oncotic pressure should have been beneficial- but we now believe that the damage caused to the glycocalyx plus the damage to the kidneys are why they aren’t as good, despite being better volume expanders.
o Saline vs balanced crystalloids is an ongoing debate and there doesn’t seem to be a conclusion coming soon, all recent studies give similar outcomes to both but there maybe an underpowered or under-demonstrated improvement in need for renal replacement therapy and less renal injury in the balanced crystalloid group.
- The suggested volume to give before considering escalating the treatment is 30ml/kg. This seems to be the point where our fluid resus has probably replaced the losses and the balance moves to start considering the consequences of ongoing fluid resuscitation.
o The evidence of this is poor and often we need to give more fluid – the question that comes with this, “are they fluid responsive?”. Basically this asks if your fluids are still working which suggests that their cardiac output is improving with the increased preload, the assumption being that they are still on the intravascularly deplete side of things.
o A recent study showed that early restrictive fluid + vasopressor was no better than a more liberal fluid policy, but more work is being done in this area.
So we have reached the point where our patient is fluid replete – around that 30ml/kg point and they are no longer fluid responsive. The principle being we have restored the intravascular volume sufficiently (resolved the distributive problem) but we are still hypotensive and tachycardic or we may still have evidence of end organ dysfunction (poor urine output or impaired cognitive function being the easiest to monitor). The next step would be to fix the other component of haemodynamic compromise – vasodilation.
A common confusion is conflating vasopressors (those that squeeze peripheral arterioles to raise systemic vascular resistance) and inotropes (which attempt to increase myocardial contractility). Asking a heart that is struggling due to sepsis, either from poor perfusion, toxic effects directly from the infection or from metabolic consequences (acidotic environment or electrolyte disturbances), is unlikely to be successful with out resolving the underlying problem so most of our treatment is focused on the vasopressor effect of the medication we use. It is worth baring in mind that a lot of the medications will have some inotropic effect in addition to the vasopressor effect.
This principle is based around the function of Blood pressure:
BP = Systemic Vascular resistance x Cardiac Output
Cardiac Output = HR x stroke volume
By looking at these principles you can understand why we have started with fluid – there is no point in squeezing an empty bag. The meaning being if the venous return is so low that the stroke volume is insignificant it is unlikely we can raise the systemic vascular resistance sufficiently to produce a good BP.
We are usually aiming for a Mean Arterial Pressure (a proxy for organ perfusion) of 65mmHg. Below are our options for vasopressors

The established practice is using peripheral pressors in emergent situations before having central venous access – the usual ones are metaraminol, phenylephrine or adrenaline. Before starting Noradrenaline, usually centrally but occasionally peripherally. Typically they are used in combination with arterial lines for direct BP measuring to titrate reliably and in real time
When norad is insufficient vasopressin is added in.
Adjuncts to support blood pressure include Corticosteroids, Sodium Bicarbonate, Insulin and Methylene Blue.
The further away from day to day practice the evidence becomes more and more absent.
Of these adjunct, the best evidence is behind corticosteroids but it remains more controversial with the potential for immunosuppression, however the general take seems to be that no one should die from sepsis without some corticosteroids.
The rationale for steroid us is fairly convincing even if the trail data isn’t quite there.
· Reversal of Relative Adrenal Insufficiency
o Hard to identify these patients that don’t have the obvious addisons features or risks (long term steroids)
· Reversal of Inflammatory Overactivity
o As we have discussed a lot of the problems caused in sepsis stem from a systemic, potentially inappropriate, inflammatory response
o Inadequate cortisol response to stress results in an increased proinflammatory state. Much of the anti-inflammatory cytokine mediation is through the glucocorticoid receptor alpha.
· Corticosteroids seems to improve vascular reactivity to Alpha-1 agonists when the response clinically has been dampened
· Can contribute to the deactivation of nitric oxide synthase – one of the major players in vasoplegia. NOS seems to be activated by both bacterial endotoxin and inflammatory cytokines
· There is some isolated heart muscle lab studies that suggests corticosteroids may offer a protective effect to sepsis induced cardiomyopathy by mitigating the effect of bacterial endotoxins on the heart
· Fluid retention
o The mild mineralocorticoid effect causes Na and then H2O retention
o Where the fluid ends up, with the leaky capillaries is a different question
· They repair/protect the Endothelial Glycocalyx
o Well accepted as a concept but the study was done in disembodied guinea pig hearts, whether you can justify it in our patients is again, another question.
From an EM perspective it may be worth considering steroids in our septic patients if they remain particularly unwell and refractory to our treatment whilst awaiting ICU involvement – the benefit being that they are drugs familiar to most of us and so a likely to be safe in our hand, where as some of the pressors may make us feel uncomfortable until we have build more familiarity. Based on the current evidence it seems that “mildly septic” patients probably don’t need steroids and the sick might but the evidence is sketchy even though the rationale is logical. This is how we have reached the “no one should die without a trial of steroid” point.
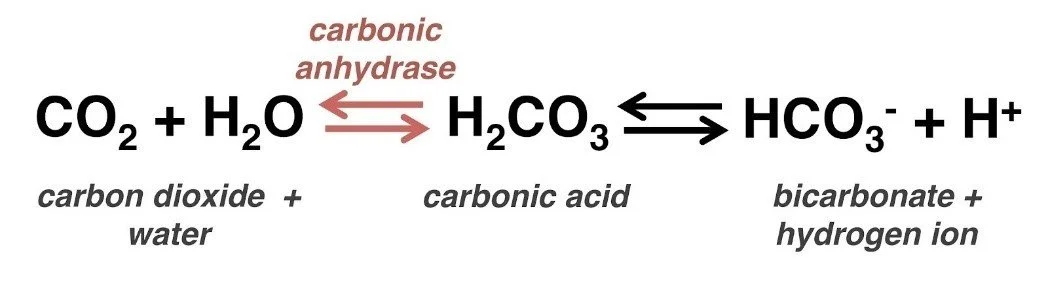
Another adjunct/Intervention that makes logical sense but has little effect in clinical trails is Sodium Bicarbonate
· As we discussed earlier the impact on myocardial contractility and function is impaired by acidosis, logically by administering NaHCO3, which acts as a buffer. We can improve the functioning
o Potential flaw in this argument – what happens when we give the HCO3.
For this question we need our Henderson-Hasselbach Equation and apply it to bicarb
If we add more HCO3 to this equation we move the equilibrium to the left, causing increased CO2 production and simply, an increase in the patient’s respiratory minute volume
o Now this limitation is born out in the trail data, where Bicarb seems to make no difference to outcome
· Now in reality, much like the steroids, it gets used in ICU when other options seem to no be working well and there is little to lose.
Now the remaining options are even more limited in their use – almost to not at all.
- Insulin is positively inotropic and increases catecholeamine sensitivity
Antibiotics
Now we haven’t really talked about antibiotics – we have focused almost solely on the supportive interventions around sepsis. The main reason for that is that antibiotic therapy should be targeted on the source and any culture results that we have.
The key, generic point about antibiotics is the timing- the earlier the better. A similar point can be applied to vasopressors, when they are needed.